Jmolを用いた研究紹介のデモ(2008年3月)
(Java Runtime Environmentのインストールが必要です)オープンキャンパス2008でのデモ内容

2008年2月18日 情報A棟5階A506の部屋でお待ちしております。
講座概要
情報科学と生物学の融合領域「バイオインフォマティクス」 において、本講座は、蛋白質の立体構造データを駆使する「構 造バイオインフォマティクス(structural bioinformatics)」の 分野を担当し、蛋白質の配列と構造との関係、さらに構造と機 能の関係の理解を目指した理論的・情報学的な研究を行います。 そのためには、データベースの単なる統計調査や既存のソフト ウエアを使用した計算機実験にとどまらず、新規のアルゴリズ ムやプログラムの開発も積極的に行います。
蛋白質の配列・構造・機能の関係を知る
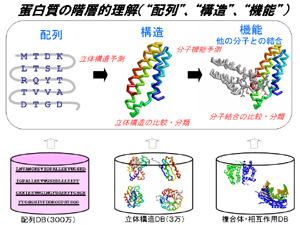 蛋白質は、生体内で様々な機能を担う、重要な分子です。
化学的には20種のアミノ酸が特定の順番で重合した高分子ですが、
その配列の並び方によって、様々な形に折り畳まり、
極めて多様な物性、機能を持つことができます。
この蛋白質という分子を特徴づけるデータとして、
20種のアミノ酸の並び方を示した「配列」、X線結晶解析や
NMRで得られた立体的な「構造」、ほかの分子のデータと結合した
複合体の立体構造、プロテオミクスなど様々な相互作用をまとめた
「機能」の3種類があり、それぞれ実験データを集めた巨大なデータベースが
世界中で作成されています。我々は、これらのデータベースを駆使し、
配列、構造、機能の関係を情報科学的に明らかにしていくことを
研究の目的としています。
蛋白質は、生体内で様々な機能を担う、重要な分子です。
化学的には20種のアミノ酸が特定の順番で重合した高分子ですが、
その配列の並び方によって、様々な形に折り畳まり、
極めて多様な物性、機能を持つことができます。
この蛋白質という分子を特徴づけるデータとして、
20種のアミノ酸の並び方を示した「配列」、X線結晶解析や
NMRで得られた立体的な「構造」、ほかの分子のデータと結合した
複合体の立体構造、プロテオミクスなど様々な相互作用をまとめた
「機能」の3種類があり、それぞれ実験データを集めた巨大なデータベースが
世界中で作成されています。我々は、これらのデータベースを駆使し、
配列、構造、機能の関係を情報科学的に明らかにしていくことを
研究の目的としています。
主な研究分野
- 蛋白質の立体構造比較と立体構造予測
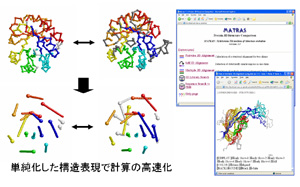 現在、3万以上の蛋白質の立体構造データが決定されており、
その構造を比較・分類することの重要性は増しています。我々
は立体構造比較プログラムMATRASを開発し、その比較サー
ビスを行うWEBサーバも立ち上げています。また、ホモロジ
ー・モデリングによる立体構造予測の改良も進めています。
現在、3万以上の蛋白質の立体構造データが決定されており、
その構造を比較・分類することの重要性は増しています。我々
は立体構造比較プログラムMATRASを開発し、その比較サー
ビスを行うWEBサーバも立ち上げています。また、ホモロジ
ー・モデリングによる立体構造予測の改良も進めています。
- 複合体のモデリングによる蛋白質間相互作用予測
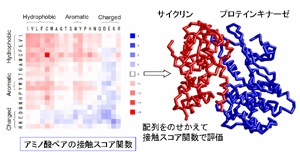 蛋白質間の相互作用は多くの生命活動の基盤をなすため、もし、
アミノ酸配列から相互作用する相手の蛋白質を絞り込むことが
できれば、その蛋白質の機能についての知見が得られます。
我々は、ゲノム中に多数のホモログが存在する蛋白質ファミリ
ー間の相互作用について、それぞれのホモログが相互作用する
かどうかを、ホモロジー・モデリングで予測された複合体立体
構造をもとに、アミノ酸ペアの接触スコア関数を計算すること
で判別しようとしています。
蛋白質間の相互作用は多くの生命活動の基盤をなすため、もし、
アミノ酸配列から相互作用する相手の蛋白質を絞り込むことが
できれば、その蛋白質の機能についての知見が得られます。
我々は、ゲノム中に多数のホモログが存在する蛋白質ファミリ
ー間の相互作用について、それぞれのホモログが相互作用する
かどうかを、ホモロジー・モデリングで予測された複合体立体
構造をもとに、アミノ酸ペアの接触スコア関数を計算すること
で判別しようとしています。
- ポケット形状の発見による低分子結合部位の推定
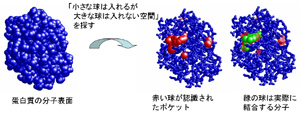 低分子はポケット形状をした表面に好んで結合することが知ら
れています。我々は、大小2種のプローブ球を用いて蛋白質表
面からポケット形状部を発見するプログラムPHECOMを新た
に開発しました。このプログラムは球の凸包のアルゴリズムを
拡張した計算幾何学のアルゴリズムを用いています。
また、同じアイデアを3次元画像処理の技術モルフォロジーを
用いて実装したプログラムGHECOMも開発しています。
これらのプログラムをもとに、より詳細な形状・原子配置を考慮した、低分
子結合部位の分類・予測を行っていきます。
低分子はポケット形状をした表面に好んで結合することが知ら
れています。我々は、大小2種のプローブ球を用いて蛋白質表
面からポケット形状部を発見するプログラムPHECOMを新た
に開発しました。このプログラムは球の凸包のアルゴリズムを
拡張した計算幾何学のアルゴリズムを用いています。
また、同じアイデアを3次元画像処理の技術モルフォロジーを
用いて実装したプログラムGHECOMも開発しています。
これらのプログラムをもとに、より詳細な形状・原子配置を考慮した、低分
子結合部位の分類・予測を行っていきます。
- 混合正規分布を用いた蛋白質構造近似表現
 多数の原子の集合である蛋白質構造を、少数の正規分布の和で
近似表現する方法を開発しています。この表現法を用いて、2
つの構造のドッキング計算、および、電子顕微鏡による3次元
電子密度に原子モデルを当てはめる計算、を高速に計算するこ
とを考えています。
多数の原子の集合である蛋白質構造を、少数の正規分布の和で
近似表現する方法を開発しています。この表現法を用いて、2
つの構造のドッキング計算、および、電子顕微鏡による3次元
電子密度に原子モデルを当てはめる計算、を高速に計算するこ
とを考えています。