Bioinformatics Unit : Go group
Research Area
Outline
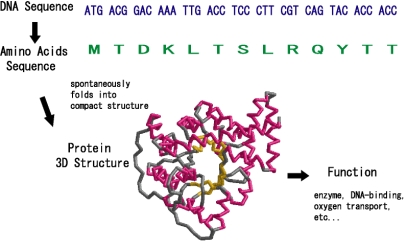
Amino acid sequences fold into compact domains with specific three-dimensional structures. These unique structural units can serve as catalytic or binding sites, as found in enzymes or DNA-binding proteins. Therefore, analysis and prediction of protein 3D structure is important for understanding the biological function of proteins. In our group we perform research in the broad area of protein 3D structure using theoretical and computational tools. The ultimate aim is to understand folding, evolution and biological functions of proteins.
Research Area
Analysis and Comparison of Protein 3D structures
A large-scale project devoted to protein structure determination, generally known as "structural genomics", was initiated recently by several organizations worldwide. As a result, it is expected that a large number of protein 3D structures will become available in the near future. Given this scenario, it becomes essential that new theoretical techniques and tools be developed for a proper analysis of the available and forthcoming 3D data for understanding protein function and evolution.
Comparison of protein 3D structures is useful for deducing their evolutionary history. Similarity of 3D structure detects more distant evolutional relationship than that of sequences, because rough patterns of 3D structures are conserved better than the corresponding sequences. We have already developed a powerful 3D structure comparison program,
"MATRAS",
which is based on Markov transition model of structure evolution. It can detect evolutionary related similarity ("homology") excluding other similarities ("analogy"), much better than other methods.

Two kinds of 3D structure similarity: "homology" and "analogy"
The detailed comparison at the atomic level is also important for a proper understanding of interactions between proteins and ligands, such as nucleotides, peptides, metal ion, and sugar. Currently we are interested in formulating general rules for ligand binding to proteins. By a proper comparison of data of protein and protein-ligand complexes, we hope to be able to deduce the function of a protein from 3D structural data alone. In addition, we are interested in identifying novel structural motifs in proteins, an example being super-secondary structural elements containing the non-canonical 310-helix.
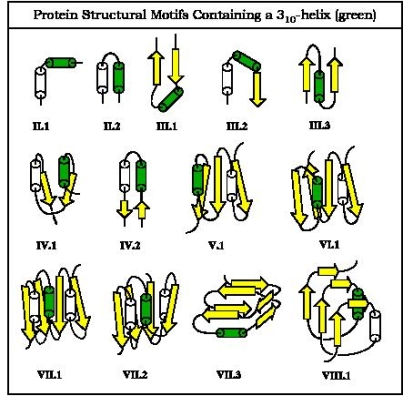
Analysis for 310 helices
Analysis of Protein Sequences
Large-scale genome projects generate enormous number of hypothetical protein sequences, their functional analyses become crucial in the post-sequence era. We attempt to analyze such sequences in the light of 3D structures.
Å@Protein 3D structure prediction from amino acid sequences is the most basic characterization of protein sequence. There are two different approaches to predict protein structures: comparative modeling and ab initio prediction. In the comparative modeling approach, one can predict 3D structure for protein sequences only if their homologs are found in the database of proteins with known structures. Ab initio prediction has no such limitation, but its accuracy is not high. Å@We aim to improve the existing prediction methods and develop new prediction methods.
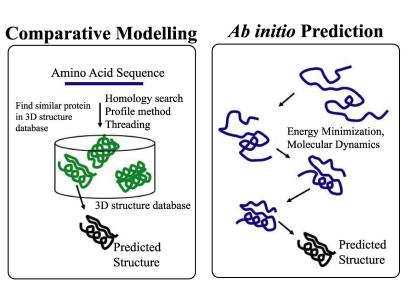
Two different approarches for structure prediction